A 9-month-old baby with subdural hematomas, retinal hemorrhages, and developmental delay
JANE F. KNAPP, MD, Section Editor; Authors: SARAH E. SODEN, MD; MAJED J. DASOUKI, MD; IRENE R. WALSH, MD
From the Section of Behavioral and Developmental Pediatrics (S.E. Soden), the Section of Medical Genetics and Molecular Medicine (M.J. Dasouki), and the Division of Emergency Medicine (I.R. Walsh), The Children´s Mercy Hospital, Kansas City, Missouri.
PEDIATRIC EMERGENCY CARE 2002;18:44-47
Key Words: Glutaric aciduria; retinal hemorrhages; subdural hematomas; developmental delay; child abuse
CASE
A 9-month-old male presented to an emergency department (ED) after collapsing in his home. His parents reported that he fell backward while kneeling on a carpeted floor at home. He struck his head on the floor, stiffened briefly, and then became limp for 5 to 10 minutes. Perioral cyanosis developed during the episode, but it resolved with 2 rescue breaths. He was transported by ambulance to the local hospital. Upon arrival in the ED, he was awake and alert. Physical examination was documented as normal. A computed tomography (CT) scan of the head was done, interpreted in the ED as normal, and the patient was discharged.
The following day, he developed vomiting and returned to the ED. A diagnosis of otitis media was made, and he was discharged. Two days later, he was admitted to the community hospital for recurrent vomiting and dehydration. A repeat CT was done, which showed a right isoattenuating (subacute) parietal subdural hematoma (Fig. 1). The next day he was noted to have less irritability and good oral intake, and was discharged. No additional studies were obtained.
JANE F. KNAPP, MD, Section Editor; Authors: SARAH E. SODEN, MD; MAJED J. DASOUKI, MD; IRENE R. WALSH, MD
From the Section of Behavioral and Developmental Pediatrics (S.E. Soden), the Section of Medical Genetics and Molecular Medicine (M.J. Dasouki), and the Division of Emergency Medicine (I.R. Walsh), The Children´s Mercy Hospital, Kansas City, Missouri.
PEDIATRIC EMERGENCY CARE 2002;18:44-47
Key Words: Glutaric aciduria; retinal hemorrhages; subdural hematomas; developmental delay; child abuse
CASE
A 9-month-old male presented to an emergency department (ED) after collapsing in his home. His parents reported that he fell backward while kneeling on a carpeted floor at home. He struck his head on the floor, stiffened briefly, and then became limp for 5 to 10 minutes. Perioral cyanosis developed during the episode, but it resolved with 2 rescue breaths. He was transported by ambulance to the local hospital. Upon arrival in the ED, he was awake and alert. Physical examination was documented as normal. A computed tomography (CT) scan of the head was done, interpreted in the ED as normal, and the patient was discharged.
The following day, he developed vomiting and returned to the ED. A diagnosis of otitis media was made, and he was discharged. Two days later, he was admitted to the community hospital for recurrent vomiting and dehydration. A repeat CT was done, which showed a right isoattenuating (subacute) parietal subdural hematoma (Fig. 1). The next day he was noted to have less irritability and good oral intake, and was discharged. No additional studies were obtained.
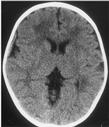
FIG. 1. A CT scan taken at the time of the patient´s first admission to the community hospital. It shows an iso-attenuating (subacute) subdural hemorrhage adjacent to the right parietal lobe.
--------------------------------------------------------------------------------
Six weeks later, while cruising along furniture, he fell again. He stiffened, developed rhythmic jerking of all extremities, and then lost consciousness for approximately 5 minutes. In the local ED, he was noted to have extreme irritability and vomiting. A head CT scan was repeated (Fig. 2). It showed bilateral hypoattenuating extraaxial fluid collections and an acute subdural hemorrhage on the right. The CT scan was also interpreted as showing concentric effacement of the ventricular system and sulci, suggestive of increased intercranial pressure. He underwent urgent craniotomy for decompression and placement of a subdural drain. Intraoperatively, bleeding vessels were cauterized, and a subdural membrane was identified.
--------------------------------------------------------------------------------
Six weeks later, while cruising along furniture, he fell again. He stiffened, developed rhythmic jerking of all extremities, and then lost consciousness for approximately 5 minutes. In the local ED, he was noted to have extreme irritability and vomiting. A head CT scan was repeated (Fig. 2). It showed bilateral hypoattenuating extraaxial fluid collections and an acute subdural hemorrhage on the right. The CT scan was also interpreted as showing concentric effacement of the ventricular system and sulci, suggestive of increased intercranial pressure. He underwent urgent craniotomy for decompression and placement of a subdural drain. Intraoperatively, bleeding vessels were cauterized, and a subdural membrane was identified.
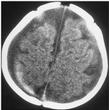
FIG. 2. A repeat CT scan 6 weeks later. Chronic subdural hematomas/hygromas are now apparent as hypo-attenuating bilateral extraaxial fluid collections. A hyper-attenuating (acute) subdural hemorrhage is also seen layering on the right.
--------------------------------------------------------------------------------
The patient was transferred to the Children´s Mercy Hospital (CMH) for further management. On detailed past medical history, his parents reported that his trunk control seemed to lag behind that of other children. He had recently begun to crawl and pull to a stand, but could not yet sit independently. There was no history of easy bruising or bleeding, and no family history of bleeding disorders. On physical examination he was sleepy but woke to painful stimuli. Pertinent findings included a prominent forehead and relative macrocephaly. Head circumference was 47.5 cm (90%) while weight and height were between the 10 and 25%. He was also noted to have truncal hypotonia. The liver edge was palpated 4 cm below the subcostal margin; the liver span was not recorded. A retinal examination revealed multiple intraretinal and 1 subhyaloid hemorrhage. Mild papilledema was also present. Hemoglobin was 10.1 g/dL. The prothrombin time (PT) was elevated at 16.3 seconds (normal 10.8-13.9 sec); however, the partial thromboplastin time (PTT), fibrinogen, and D-dimers were all within normal limits. His blood chemistry showed a CO2 of 14 mmol/L with an anion gap of 15 mmol/L. A skeletal survey demonstrated no fractures.
DIFFERENTIAL DIAGNOSIS
At the time of presentation to CMH, this patient had evidence of both chronic and acute subdural bleeding, as well as retinal hemorrhages. One or more falls from standing or kneeling in an otherwise healthy child would not result in such findings (1,2). Identification of a subdural hemorrhage necessitates further evaluation. A thorough examination for possible child abuse is indicated, as is a careful consideration of underlying medical conditions that increase a child´s susceptibility to intracranial bleeding.
Malignancies, infections, and coagulation disorders are among the medical conditions to consider. Our patient did have a mildly elevated prothrombin time when he arrived at CMH, but it had previously been normal, and he had no other findings consistent with a bleeding diathesis. It is important to remember that release of cerebral thromboplastin can cause a secondary coagulopathy in some patients with cerebral injury. Additional laboratory work, including the measurement of individual clotting factors, may be necessary if the diagnosis is uncertain.
Duhaime described another group of patients with increased vulnerability to subdural hemorrhages in 1992. Patients with enlarged extraaxial spaces, such as those with shunted hydrocephalus or cortical atrophy, may have stretching of their bridging veins across these spaces, making the patients susceptible to profuse bleeding caused by relatively small acceleration forces (3).
A high index of suspicion for inborn errors of metabolism is necessary when evaluating infants with unusual or unexplained deterioration. Glutaric aciduria type 1 is an autosomal recessive condition, which typically presents in the 1st or 2nd year of life. Subdural hematomas and retinal hemorrhages have been reported in some cases (4). The risk of subdural hematomas and effusions in affected patients is estimated at 20 to 30% (5). Ocular findings, including hemorrhages and cataracts, were discovered in more than 40% of patients in a recent series (6). Increased intracranial pressure and alterations in protein synthesis or structure of the lens membrane were proposed as possible etiologies.
The leading cause of serious head injury in infants is child abuse (1). Evaluation for abuse must not be delayed during investigation for other underlying medical conditions. Thorough histories should be taken while the events are still fresh in the minds of the child´s caretakers. A skeletal survey, ophthalmology examination, and careful inspection of the integument are essential components of the early management of young children presenting with subdural hematomas of uncertain etiology. In most cases, hospitalization and referral to child protection services for further investigation is warranted.
Differentiation of retinal hemorrhages caused by shaking/impact syndrome from those caused by other mechanisms can be challenging. Numerous hemorrhages involving all layers of the retina and extending to the periphery are characteristic of intentional trauma (7). The presence of retinal folds and retinoschisis is also strongly suggestive (Fig. 3). Retinal photography is valuable for documentation and review.
--------------------------------------------------------------------------------
The patient was transferred to the Children´s Mercy Hospital (CMH) for further management. On detailed past medical history, his parents reported that his trunk control seemed to lag behind that of other children. He had recently begun to crawl and pull to a stand, but could not yet sit independently. There was no history of easy bruising or bleeding, and no family history of bleeding disorders. On physical examination he was sleepy but woke to painful stimuli. Pertinent findings included a prominent forehead and relative macrocephaly. Head circumference was 47.5 cm (90%) while weight and height were between the 10 and 25%. He was also noted to have truncal hypotonia. The liver edge was palpated 4 cm below the subcostal margin; the liver span was not recorded. A retinal examination revealed multiple intraretinal and 1 subhyaloid hemorrhage. Mild papilledema was also present. Hemoglobin was 10.1 g/dL. The prothrombin time (PT) was elevated at 16.3 seconds (normal 10.8-13.9 sec); however, the partial thromboplastin time (PTT), fibrinogen, and D-dimers were all within normal limits. His blood chemistry showed a CO2 of 14 mmol/L with an anion gap of 15 mmol/L. A skeletal survey demonstrated no fractures.
DIFFERENTIAL DIAGNOSIS
At the time of presentation to CMH, this patient had evidence of both chronic and acute subdural bleeding, as well as retinal hemorrhages. One or more falls from standing or kneeling in an otherwise healthy child would not result in such findings (1,2). Identification of a subdural hemorrhage necessitates further evaluation. A thorough examination for possible child abuse is indicated, as is a careful consideration of underlying medical conditions that increase a child´s susceptibility to intracranial bleeding.
Malignancies, infections, and coagulation disorders are among the medical conditions to consider. Our patient did have a mildly elevated prothrombin time when he arrived at CMH, but it had previously been normal, and he had no other findings consistent with a bleeding diathesis. It is important to remember that release of cerebral thromboplastin can cause a secondary coagulopathy in some patients with cerebral injury. Additional laboratory work, including the measurement of individual clotting factors, may be necessary if the diagnosis is uncertain.
Duhaime described another group of patients with increased vulnerability to subdural hemorrhages in 1992. Patients with enlarged extraaxial spaces, such as those with shunted hydrocephalus or cortical atrophy, may have stretching of their bridging veins across these spaces, making the patients susceptible to profuse bleeding caused by relatively small acceleration forces (3).
A high index of suspicion for inborn errors of metabolism is necessary when evaluating infants with unusual or unexplained deterioration. Glutaric aciduria type 1 is an autosomal recessive condition, which typically presents in the 1st or 2nd year of life. Subdural hematomas and retinal hemorrhages have been reported in some cases (4). The risk of subdural hematomas and effusions in affected patients is estimated at 20 to 30% (5). Ocular findings, including hemorrhages and cataracts, were discovered in more than 40% of patients in a recent series (6). Increased intracranial pressure and alterations in protein synthesis or structure of the lens membrane were proposed as possible etiologies.
The leading cause of serious head injury in infants is child abuse (1). Evaluation for abuse must not be delayed during investigation for other underlying medical conditions. Thorough histories should be taken while the events are still fresh in the minds of the child´s caretakers. A skeletal survey, ophthalmology examination, and careful inspection of the integument are essential components of the early management of young children presenting with subdural hematomas of uncertain etiology. In most cases, hospitalization and referral to child protection services for further investigation is warranted.
Differentiation of retinal hemorrhages caused by shaking/impact syndrome from those caused by other mechanisms can be challenging. Numerous hemorrhages involving all layers of the retina and extending to the periphery are characteristic of intentional trauma (7). The presence of retinal folds and retinoschisis is also strongly suggestive (Fig. 3). Retinal photography is valuable for documentation and review.
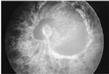
FIG. 3. A retinal photograph of a victim of shaking/impact syndrome showing diffuse, multilayered retinal hemorrhages and a retinal fold.
--------------------------------------------------------------------------------
HOSPITAL COURSE
The patient was admitted to the pediatric intensive care unit, and the Child Protection Team was consulted. Their recommendations included screening for glutaric aciduria type 1 (GA1). Metabolic serum and urine studies were obtained. The patient was fully awake, and stable for transfer to the pediatric ward by the following day. Two liters of bloody fluid drained from the subdural catheter during the first week. His metabolic studies returned during the second week of hospitalization. Urine organic acid testing showed elevated excretion of 3-hydroxyglutaric acid and glutaric acid (4583 mmol/mol creatanine, normal <10 mmol/mol). These findings, along with reduced serum free and acyl carnitine, are characteristic of GA1.
Appropriate dietary management was initiated, including carnitine and riboflavin supplements, and a diet with restricted lysine and tryptophan. His hospital course was complicated by an infection of the subdural drain. The drain was removed, but subdural effusions reaccumulated, and the patient developed increased intracranial pressure. He ultimately required placement of a subdural-peritoneal shunt. The family received extensive education on the management of patients with GA1. Importantly, prior to their child´s discharge, they were given a written plan for acute management of subsequent illnesses or metabolic crises that could be provided to health care workers unfamiliar with GA1.
His illness was complicated by poor oral intake, making gastrostomy tube placement necessary. Challenging episodes of irritability, seizures, and severe dystonia developed. Dystonia did not respond to treatment with carbidopa-levodopa (Sinemet; DuPont Merck Pharmaceutical, Wilmington, DE) or baclofen. Direct intramuscular injections of botulinum toxin resulted in partial relief of the painful dystonic posturing. Ultimately, he underwent a neurosurgical procedure, bilateral pallidotomy, to control his dystonia.
DISCUSSION
Glutaric aciduria type 1, an autosomal recessive inborn error of metabolism, was first described by Goodman, et al. in 1975 (8). Affected individuals have abnormal metabolism of lysine, tryptophan, and hydroxylysine due to a deficiency of the mitochondrial enzyme glutaryl-CoA dehydrogenase. Glutaryl-CoA accumulates proximal to the enzymatic block, and is hydrolyzed to glutaric acid. The enzyme has been mapped to chromosome 19p13.2 (9) and the incidence has been estimated at 1 in 30,000 (10). Affected individuals typically have normal or near normal development in early infancy. Subtle early findings, such as truncal hypotonia and relative macrocephaly as seen in this patient, are often present but easily overlooked. Patients frequently suffer an episode of metabolic decompensation during their first or second year of life. The mild hypovolemia, hypoglycemia, and fever tolerated by most infants and toddlers during routine childhood illnesses can be devastating to these patients. They develop a catabolic state with accumulation of metabolic byproducts resulting in neurotoxicity. Some children present with a clinical picture common to other organic acidemias with hypoglycemia, acidosis, and hyperammonemia. However, many will not have acidosis (4). Affected individuals may present with unexplained altered mental status, atypical movements, seizures, or irritability. Classically, patients develop dystonia and athetosis with relative sparing of intellect (11). A subset of individuals has a more insidious onset and some remain asymptomatic into adulthood (14,20). Historically, misdiagnoses have included infectious encephalopathy, athetoid cerebral palsy, Reye syndrome, and shaking/impact syndrome (19,21,22).
Elevated glutaric acid can be detected in the blood, urine, cerebrospinal fluid (CSF), and tissues (12). Patients have a secondary carnitine deficiency, which is detectable with serum testing. Glutaric acid has been shown to have a direct toxic effect on striatal cells in vitro (13). Concentrations of -aminobutyric acid are low in the basal ganglia of these patients, and the metabolism of -aminobutyric acid is affected by glutaric acid (14). Quinolinic acid, an intermediary of tryptophan metabolism, is another possible source of neurotoxicity (15). The caudate and putamen shrink markedly in response to metabolic insult. Histologically, neuronal loss and gliosis are seen (16). Fatty changes of the liver, heart, and kidneys have been described on postmortem examination and may be secondary to mitochondrial toxicity (17).
A number of radiologic findings are characteristic of GA1. Changes on CT scan can be present even prior to the 1st metabolic crisis, and are an important early diagnostic clue (18). Frontotemporal atrophy, subdural fluid collections, widened CSF spaces, particularly wide sylvian fissures, and dilated insular cisterns are frequently described on CT scan or MRI (18,19). Progressive cortical atrophy, white matter attenuation, and basal ganglia changes appear with disease progression. On MRI, basal ganglia changes typically begin with increased T2 signal and progress to loss of volume, particularly of the caudate and putamen (14).
There is compelling evidence that dietary management and prevention of a catabolic state during illness reduce morbidity and mortality (12,19). Ideally, a low lysine and tryptophan diet with carnitine and riboflavin supplements should be started in infancy. Irritability, anorexia, vomiting, lethargy, and worsening hypotonia are signs of metabolic decompensation in these patients and should be considered a metabolic emergency. The child may appear toxic or encephalopathic. Anion gap acidosis may be present. Glucose should be administered at twice the patient´s basal metabolic rate. Administration of insulin may also be necessary during the glucose infusion to maintain blood glucose between 100 and 150 mg/dL. Recovery may depend on the renal clearance of non-volatile dicarboxylic acids. This requires intravascular volume expansion to establish vigorous urine output. A urine pH greater than 7.5 has been recommended, as well as close attention to maintenance of a normal serum sodium and potassium. In cases in which profound acidosis persists despite volume expansion and glucose infusion, treatment with sodium bicarbonate can be considered.
Differentiation between GA1 and intentional trauma may be extremely difficult. In our case, hypotonia, an atypical developmental history, and accelerated head growth were important clues. The early hospital course at CMH was also atypical for that of a shaken baby. For example, this patient had extremely large subdural fluid collections, yet he was awake and alert by the second hospital day. In anatomically normal brains, large subdural hemorrhages are often the result of angular acceleration of the brain. This results in severe shearing of bridging vessels and shearing forces on the brain parenchyma. This type of parenchymal damage is known as diffuse axonal injury and causes profound neurologic sequelae and can be fatal (3,23).
Premorbid signs of GA1 may be subtle or nonexistent, and some authors have argued that all infants being evaluated for the shaking/impact syndrome should be screened for GA1. While there is an increasing awareness of GA1 among child protection experts, there is no consensus regarding screening at this time.
CONCLUSIONS
Infants who present with unexplained subdural and retinal hemorrhages most often are victims of child abuse. However, an autosomal recessive metabolic disorder, glutaric aciduria type 1, is a known cause of these findings in children. Urine organic acid testing will show a characteristic large peak of glutaric acid and 3hydroxy glutaric acid. The diagnosis can be confirmed through testing of enzyme activity in cultured fibroblasts and leukocytes or through a genetic mutation analysis. Early recognition and treatment of this disorder has the potential for reducing the morbidity and mortality associated with GA1.
REFERENCES
Committee on Child Abuse and Neglect. Shaken baby syndrome: Rotational cranial injuries-technical report (T0039). Pediatrics 2001;108:206-210.
Duhaime AC, Christian CW, Rorke LB, et al. Nonaccidental head injury in infants - The "shaken baby syndrome." N Engl J Med 1998;338:1822-1829.
Duhaime AC, Alario AJ, Lewander WJ, et al. Head injury in very young children: Mechanisms, injury types, and opthamologic findings in 100 hospitalized patients less than 2 years of age. Pediatrics 1992;90:179-185.
Hartley LM, Khwaja OS, Verity CM. Glutaric aciduria type 1 and nonaccidental head injury. Pediatrics 2001;107:174-175.
Hoffmann GF, Bohles HJ, Burlina A, et al. Early signs and course of disease of glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 1995;18:173-176.
Kafil-Hussain NA, Monavari A, Bowell R, et al. Ocular findings in glutaric aciduria type 1. J Pediatr Ophthalmol Strabismus 2000;37:289-293.
Christian CW, Taylor AA, Hertle RW, et al. Retinal hemorrhages caused by accidental household trauma. J Pediatr 1999;135:125-127.
Goodman SI, Markey SP, Moe PG, et al. Glutaric aciduria: A "new" disorder of amino acid metabolism. Biochem Med 1975;12:12-21.
Greenberg CR, Duncan AM, Gregory CA, et al. Assignment of human glutaryl-CoA dehydrogenase gene (GCDH) to the short arm of chromosome 19 (19p13.2) by in situ hybridization and somatic cell hybrid analysis. Genomics 1994;21:289-290.
Kyllerman M, Steen G. Glutaric aciduria: A "common" metabolic disorder? Arch Fr Pediatr 1980;37:279.
Morton DH, Bennett MJ, Seargeant LE, et al. Glutaric aciduria type 1: A common cause of episodic encephalopathy and spastic paralysis in the Amish of Lancaster County, Pennsylvania. Am J Med Genet 1991;41:89-95.
Goodman SI, Frerman FE. Organic acidemias due to defects in lysine oxidation: 2-ketoadipic acidemia and glutaric acidemia. In: Scriver CR, et al. eds: The metabolic basis of inherited disease, ed 7. New York: McGraw Hill; 1995:1451-1460.
Whetsell Jr. WO The use of organotypic tissue culture for study of amino acid neurotoxicity and its antagonism in mammalian CNS. Clin Neuropharmacol 1984;7(suppl 1):452-453.
Amir N, el-Peleg O, Shalev RS, et al. Glutaric aciduria type 1: Clinical heterogeneity and neuroradiologic features. Neurology 1987;37:1654-1657.
McGeer EG, Singh E. Neurotoxic effects of endogenous materials: Quinolinic acid, L-pyroglutamic acid, and thyroid releasing hormone. Exp Neurol 1984;86:410-413.
Goodman SI, Norenberg MD, Shikes RH, et al. Glutaric acidurea: Biochemical and morphologic considerations. J Pediatr 1977;90:746-750.
Gregersen N, Brandt NJ. Ketotic episodes in glutaryl-CoA dehydrogenase deficiency. Pediatr Res 1979;13:997-981.
Brismar J, Ozand PT. CT and MR in the brain of glutamic acidemia type 1: A review of 59 published cases and report of 5 new patients. Am J Neuroradiol 1995;16:675-683.
Yager JY, McClarty BM, Seshia SS. CT-scan findings in an infant with glutaric aciduria type 1. Dev Med Child Neurol 1988;30:808-811.
Haworth JC, Booth FA, Chudley AE, et al. Phenotypic variability in glutaric aciduria type 1: Report of fourteen cases in five Canadian Indian kindreds. J Pediatr 1991;118:52-58.
Bennett MJ, Marlow N, Politt RJ, et al. Glutaric aciduria type 1: Biochemical investigations and postmortem findings. Eur J Pediatr 1986; 145:403-405.
Brandt NJ, Brandt S, Christensen E, et al. Glutaric aciduria in progressive choreo-athetosis. Clin Genet 1978;13:77-80.
Hymel KP, Bandak FA, Partington MD, et al. Abusive head trauma? A biomechanics-based approach. Child Maltreat 1998;3:116-128.
Address for reprints: Sarah Soden, MD, Section of Behavioral and Developmental Pediatrics, The Children´s Mercy Hospital, 2401 Gillham Road, Kansas City, MO 64108; e-mail: ssoden@cmh.edu
The authors would like to acknowledge Drs. M. Stass-Isern and T. Hug for the retinal photograph, Drs. R. Gravis and L. Lowe for their assistance with the radiology, and Dr. M. Tuchman for performing the initial urine organic analysis on this patient.
Pediatr Emerg Care 2002 February;18(1):44-47
Copyright © 2002 Lippincott Williams & Wilkins
All rights reserved
--------------------------------------------------------------------------------
HOSPITAL COURSE
The patient was admitted to the pediatric intensive care unit, and the Child Protection Team was consulted. Their recommendations included screening for glutaric aciduria type 1 (GA1). Metabolic serum and urine studies were obtained. The patient was fully awake, and stable for transfer to the pediatric ward by the following day. Two liters of bloody fluid drained from the subdural catheter during the first week. His metabolic studies returned during the second week of hospitalization. Urine organic acid testing showed elevated excretion of 3-hydroxyglutaric acid and glutaric acid (4583 mmol/mol creatanine, normal <10 mmol/mol). These findings, along with reduced serum free and acyl carnitine, are characteristic of GA1.
Appropriate dietary management was initiated, including carnitine and riboflavin supplements, and a diet with restricted lysine and tryptophan. His hospital course was complicated by an infection of the subdural drain. The drain was removed, but subdural effusions reaccumulated, and the patient developed increased intracranial pressure. He ultimately required placement of a subdural-peritoneal shunt. The family received extensive education on the management of patients with GA1. Importantly, prior to their child´s discharge, they were given a written plan for acute management of subsequent illnesses or metabolic crises that could be provided to health care workers unfamiliar with GA1.
His illness was complicated by poor oral intake, making gastrostomy tube placement necessary. Challenging episodes of irritability, seizures, and severe dystonia developed. Dystonia did not respond to treatment with carbidopa-levodopa (Sinemet; DuPont Merck Pharmaceutical, Wilmington, DE) or baclofen. Direct intramuscular injections of botulinum toxin resulted in partial relief of the painful dystonic posturing. Ultimately, he underwent a neurosurgical procedure, bilateral pallidotomy, to control his dystonia.
DISCUSSION
Glutaric aciduria type 1, an autosomal recessive inborn error of metabolism, was first described by Goodman, et al. in 1975 (8). Affected individuals have abnormal metabolism of lysine, tryptophan, and hydroxylysine due to a deficiency of the mitochondrial enzyme glutaryl-CoA dehydrogenase. Glutaryl-CoA accumulates proximal to the enzymatic block, and is hydrolyzed to glutaric acid. The enzyme has been mapped to chromosome 19p13.2 (9) and the incidence has been estimated at 1 in 30,000 (10). Affected individuals typically have normal or near normal development in early infancy. Subtle early findings, such as truncal hypotonia and relative macrocephaly as seen in this patient, are often present but easily overlooked. Patients frequently suffer an episode of metabolic decompensation during their first or second year of life. The mild hypovolemia, hypoglycemia, and fever tolerated by most infants and toddlers during routine childhood illnesses can be devastating to these patients. They develop a catabolic state with accumulation of metabolic byproducts resulting in neurotoxicity. Some children present with a clinical picture common to other organic acidemias with hypoglycemia, acidosis, and hyperammonemia. However, many will not have acidosis (4). Affected individuals may present with unexplained altered mental status, atypical movements, seizures, or irritability. Classically, patients develop dystonia and athetosis with relative sparing of intellect (11). A subset of individuals has a more insidious onset and some remain asymptomatic into adulthood (14,20). Historically, misdiagnoses have included infectious encephalopathy, athetoid cerebral palsy, Reye syndrome, and shaking/impact syndrome (19,21,22).
Elevated glutaric acid can be detected in the blood, urine, cerebrospinal fluid (CSF), and tissues (12). Patients have a secondary carnitine deficiency, which is detectable with serum testing. Glutaric acid has been shown to have a direct toxic effect on striatal cells in vitro (13). Concentrations of -aminobutyric acid are low in the basal ganglia of these patients, and the metabolism of -aminobutyric acid is affected by glutaric acid (14). Quinolinic acid, an intermediary of tryptophan metabolism, is another possible source of neurotoxicity (15). The caudate and putamen shrink markedly in response to metabolic insult. Histologically, neuronal loss and gliosis are seen (16). Fatty changes of the liver, heart, and kidneys have been described on postmortem examination and may be secondary to mitochondrial toxicity (17).
A number of radiologic findings are characteristic of GA1. Changes on CT scan can be present even prior to the 1st metabolic crisis, and are an important early diagnostic clue (18). Frontotemporal atrophy, subdural fluid collections, widened CSF spaces, particularly wide sylvian fissures, and dilated insular cisterns are frequently described on CT scan or MRI (18,19). Progressive cortical atrophy, white matter attenuation, and basal ganglia changes appear with disease progression. On MRI, basal ganglia changes typically begin with increased T2 signal and progress to loss of volume, particularly of the caudate and putamen (14).
There is compelling evidence that dietary management and prevention of a catabolic state during illness reduce morbidity and mortality (12,19). Ideally, a low lysine and tryptophan diet with carnitine and riboflavin supplements should be started in infancy. Irritability, anorexia, vomiting, lethargy, and worsening hypotonia are signs of metabolic decompensation in these patients and should be considered a metabolic emergency. The child may appear toxic or encephalopathic. Anion gap acidosis may be present. Glucose should be administered at twice the patient´s basal metabolic rate. Administration of insulin may also be necessary during the glucose infusion to maintain blood glucose between 100 and 150 mg/dL. Recovery may depend on the renal clearance of non-volatile dicarboxylic acids. This requires intravascular volume expansion to establish vigorous urine output. A urine pH greater than 7.5 has been recommended, as well as close attention to maintenance of a normal serum sodium and potassium. In cases in which profound acidosis persists despite volume expansion and glucose infusion, treatment with sodium bicarbonate can be considered.
Differentiation between GA1 and intentional trauma may be extremely difficult. In our case, hypotonia, an atypical developmental history, and accelerated head growth were important clues. The early hospital course at CMH was also atypical for that of a shaken baby. For example, this patient had extremely large subdural fluid collections, yet he was awake and alert by the second hospital day. In anatomically normal brains, large subdural hemorrhages are often the result of angular acceleration of the brain. This results in severe shearing of bridging vessels and shearing forces on the brain parenchyma. This type of parenchymal damage is known as diffuse axonal injury and causes profound neurologic sequelae and can be fatal (3,23).
Premorbid signs of GA1 may be subtle or nonexistent, and some authors have argued that all infants being evaluated for the shaking/impact syndrome should be screened for GA1. While there is an increasing awareness of GA1 among child protection experts, there is no consensus regarding screening at this time.
CONCLUSIONS
Infants who present with unexplained subdural and retinal hemorrhages most often are victims of child abuse. However, an autosomal recessive metabolic disorder, glutaric aciduria type 1, is a known cause of these findings in children. Urine organic acid testing will show a characteristic large peak of glutaric acid and 3hydroxy glutaric acid. The diagnosis can be confirmed through testing of enzyme activity in cultured fibroblasts and leukocytes or through a genetic mutation analysis. Early recognition and treatment of this disorder has the potential for reducing the morbidity and mortality associated with GA1.
REFERENCES
Committee on Child Abuse and Neglect. Shaken baby syndrome: Rotational cranial injuries-technical report (T0039). Pediatrics 2001;108:206-210.
Duhaime AC, Christian CW, Rorke LB, et al. Nonaccidental head injury in infants - The "shaken baby syndrome." N Engl J Med 1998;338:1822-1829.
Duhaime AC, Alario AJ, Lewander WJ, et al. Head injury in very young children: Mechanisms, injury types, and opthamologic findings in 100 hospitalized patients less than 2 years of age. Pediatrics 1992;90:179-185.
Hartley LM, Khwaja OS, Verity CM. Glutaric aciduria type 1 and nonaccidental head injury. Pediatrics 2001;107:174-175.
Hoffmann GF, Bohles HJ, Burlina A, et al. Early signs and course of disease of glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 1995;18:173-176.
Kafil-Hussain NA, Monavari A, Bowell R, et al. Ocular findings in glutaric aciduria type 1. J Pediatr Ophthalmol Strabismus 2000;37:289-293.
Christian CW, Taylor AA, Hertle RW, et al. Retinal hemorrhages caused by accidental household trauma. J Pediatr 1999;135:125-127.
Goodman SI, Markey SP, Moe PG, et al. Glutaric aciduria: A "new" disorder of amino acid metabolism. Biochem Med 1975;12:12-21.
Greenberg CR, Duncan AM, Gregory CA, et al. Assignment of human glutaryl-CoA dehydrogenase gene (GCDH) to the short arm of chromosome 19 (19p13.2) by in situ hybridization and somatic cell hybrid analysis. Genomics 1994;21:289-290.
Kyllerman M, Steen G. Glutaric aciduria: A "common" metabolic disorder? Arch Fr Pediatr 1980;37:279.
Morton DH, Bennett MJ, Seargeant LE, et al. Glutaric aciduria type 1: A common cause of episodic encephalopathy and spastic paralysis in the Amish of Lancaster County, Pennsylvania. Am J Med Genet 1991;41:89-95.
Goodman SI, Frerman FE. Organic acidemias due to defects in lysine oxidation: 2-ketoadipic acidemia and glutaric acidemia. In: Scriver CR, et al. eds: The metabolic basis of inherited disease, ed 7. New York: McGraw Hill; 1995:1451-1460.
Whetsell Jr. WO The use of organotypic tissue culture for study of amino acid neurotoxicity and its antagonism in mammalian CNS. Clin Neuropharmacol 1984;7(suppl 1):452-453.
Amir N, el-Peleg O, Shalev RS, et al. Glutaric aciduria type 1: Clinical heterogeneity and neuroradiologic features. Neurology 1987;37:1654-1657.
McGeer EG, Singh E. Neurotoxic effects of endogenous materials: Quinolinic acid, L-pyroglutamic acid, and thyroid releasing hormone. Exp Neurol 1984;86:410-413.
Goodman SI, Norenberg MD, Shikes RH, et al. Glutaric acidurea: Biochemical and morphologic considerations. J Pediatr 1977;90:746-750.
Gregersen N, Brandt NJ. Ketotic episodes in glutaryl-CoA dehydrogenase deficiency. Pediatr Res 1979;13:997-981.
Brismar J, Ozand PT. CT and MR in the brain of glutamic acidemia type 1: A review of 59 published cases and report of 5 new patients. Am J Neuroradiol 1995;16:675-683.
Yager JY, McClarty BM, Seshia SS. CT-scan findings in an infant with glutaric aciduria type 1. Dev Med Child Neurol 1988;30:808-811.
Haworth JC, Booth FA, Chudley AE, et al. Phenotypic variability in glutaric aciduria type 1: Report of fourteen cases in five Canadian Indian kindreds. J Pediatr 1991;118:52-58.
Bennett MJ, Marlow N, Politt RJ, et al. Glutaric aciduria type 1: Biochemical investigations and postmortem findings. Eur J Pediatr 1986; 145:403-405.
Brandt NJ, Brandt S, Christensen E, et al. Glutaric aciduria in progressive choreo-athetosis. Clin Genet 1978;13:77-80.
Hymel KP, Bandak FA, Partington MD, et al. Abusive head trauma? A biomechanics-based approach. Child Maltreat 1998;3:116-128.
Address for reprints: Sarah Soden, MD, Section of Behavioral and Developmental Pediatrics, The Children´s Mercy Hospital, 2401 Gillham Road, Kansas City, MO 64108; e-mail: ssoden@cmh.edu
The authors would like to acknowledge Drs. M. Stass-Isern and T. Hug for the retinal photograph, Drs. R. Gravis and L. Lowe for their assistance with the radiology, and Dr. M. Tuchman for performing the initial urine organic analysis on this patient.
Pediatr Emerg Care 2002 February;18(1):44-47
Copyright © 2002 Lippincott Williams & Wilkins
All rights reserved
